Combining multiple tibbles with dplyr
Agenda
- Combining tibbles (i.e. data joins)
- Applications of data joins to forestry
Data Joins in R
Today, we’ll talk about joining tibbles in R with dplyr.

Artwork by Allison Horst.
Combining tibbles
Data Joins in R
Recall that the dplyr package, short for “data pliers”, is an R package all about wrangling data.

Today, we will explore it’s tools for joining data tables (i.e. “tibbles”).
Data Joins in R
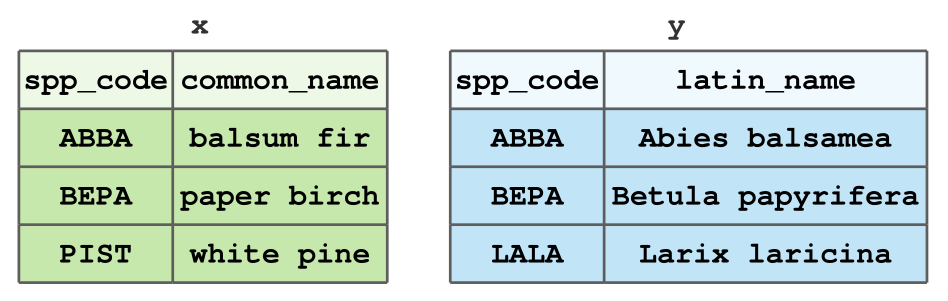
In particular, we’ll look at a few ways to join the following tables x and y:
Motivation
- It is common in forestry, and in particular forest inventory, to have multiple tibbles where data are stored due to a variety of factors.
- In order to perform most statistical analyses, you must have the necessary data in one tibble.
- Example: the US Forest Service, Forest Inventory & Analysis stores plot-level, tree-level, subplot-level, … data in one database. Researchers must combine these data into a singular table to do analyses.
Types of Data Joins
The dplyr package, which is part of the tidyverse, includes functions for two general types of joins:
- Mutating joins, which combine the columns of tibbles
xandy, and - Filtering joins, which match the rows of tibbles
xandy.
Types of Data Joins
The dplyr package, which is part of the tidyverse, includes functions for two general types of joins:
- Mutating joins, which combine the columns of tibbles
xandy, and - Filtering joins, which match the rows of tibbles
xandy.
Think of how mutate() adds columns to a tibble, while filter() removes rows.
Example Data
For the following examples of data joins, we will use the tibbles from the first slide. We can load this data into R:
Mutating Joins
dplyr contains four mutating joins:
left_join(x, y)keeps all rows ofx, but if a row inydoes not match tox, anNAis assigned to that row in the new columns.
Mutating Joins
dplyr contains four mutating joins:
left_join(x, y)keeps all rows ofx, but if a row inydoes not match tox, anNAis assigned to that row in the new columns.right_join(x, y)is equivalent toleft_join(y, x), except for column order.
Mutating Joins
dplyr contains four mutating joins:
left_join(x, y)keeps all rows ofx, but if a row inydoes not match tox, anNAis assigned to that row in the new columns.right_join(x, y)is equivalent toleft_join(y, x), except for column order.
inner_join(x, y)keeps only the rows matched betweenxandy.
Mutating Joins
dplyr contains four mutating joins:
left_join(x, y)keeps all rows ofx, but if a row inydoes not match tox, anNAis assigned to that row in the new columns.right_join(x, y)is equivalent toleft_join(y, x), except for column order.
inner_join(x, y)keeps only the rows matched betweenxandy.full_join(x, y)keeps all rows of bothxandy.
Examples: Mutating Joins
Recall our example data
left_join()
left_join()
Joining with `by = join_by(spp_code)`# A tibble: 3 × 3
spp_code common_name latin_name
<chr> <chr> <chr>
1 ABBA balsum fir Abies balsamea
2 BEPA paper birch Betula papyrifera
3 PIST white pine <NA> But what’s that message about?
"Joining with `by = join_by(spp_code)`"We need to specify a key 🔑
A key is can just be thought of the name(s) of the column(s) you’re joining by. In the left_join() from the last slide, R assumed we were joining by the column spp_code since both x and y have a column with that name.
We need to specify a key 🔑
A key is can just be thought of the name(s) of the column(s) you’re joining by. In the left_join() from the last slide, R assumed we were joining by the column spp_code since both x and y have a column with that name.
Keys are important, especially when the columns you are joining by have different names, or you are joining by multiple columns.
left_join(), with a 🔑
# A tibble: 3 × 3
spp_code common_name latin_name
<chr> <chr> <chr>
1 ABBA balsum fir Abies balsamea
2 BEPA paper birch Betula papyrifera
3 PIST white pine <NA> - Notice that we specify this key with the
byargument. This is the same for all joins indplyr.
right_join()
right_join()
# A tibble: 3 × 3
spp_code latin_name common_name
<chr> <chr> <chr>
1 ABBA Abies balsamea balsum fir
2 BEPA Betula papyrifera paper birch
3 PIST <NA> white pine Notice that this is the same as our previous left_join().
What happens if we try switching the order of x and y?
right_join()
# A tibble: 3 × 3
spp_code latin_name common_name
<chr> <chr> <chr>
1 ABBA Abies balsamea balsum fir
2 BEPA Betula papyrifera paper birch
3 PIST <NA> white pine Notice that this is the same as our previous left_join().
What happens if we try switching the order of x and y?
inner_join()
How many rows will the output have?
inner_join()
How many rows will the output have?
# A tibble: 2 × 3
spp_code common_name latin_name
<chr> <chr> <chr>
1 ABBA balsum fir Abies balsamea
2 BEPA paper birch Betula papyriferaWhy is this the result?
full_join()
How many rows will the output have?
full_join()
How many rows will the output have?
# A tibble: 4 × 3
spp_code common_name latin_name
<chr> <chr> <chr>
1 ABBA balsum fir Abies balsamea
2 BEPA paper birch Betula papyrifera
3 PIST white pine <NA>
4 LALA <NA> Larix laricina Why is this the result?
Filtering Joins
dplyr contains two filtering joins:
semi_join(x, y)keeps all the rows inxthat have a match iny.
Filtering Joins
dplyr contains two filtering joins:
semi_join(x, y)keeps all the rows inxthat have a match iny.anti_join(x, y)removes all the rows inxthat have a match iny.
Note: Unlike mutating joins, filtering joins do not add any columns to the data.
semi_join()
How many rows will this semi_join return? How many columns?
semi_join()
How many rows will this semi_join return? How many columns?
semi_join()
What about this semi_join? Will it be the same as semi_join(x, y)?
semi_join()
What about this semi_join? Will it be the same as semi_join(x, y)?
anti_join()
Let’s see what anti_join does:
anti_join()
Let’s see what anti_join does:
# A tibble: 1 × 2
spp_code common_name
<chr> <chr>
1 PIST white pine Why do we get this output?
anti_join()
What happens if we switch the order of x and y?
anti_join()
What happens if we switch the order of x and y?
An Important Subtlety: Column Names
So far, we have joined x and y by the spp_code column.
But what if y had the same column named differently:
How Do We Join x and y?
Error in `left_join()`:
! `by` must be supplied when `x` and `y` have no common variables.
ℹ Use `cross_join()` to perform a cross-join.Looks like we need to specify by (our 🔑)
How Do We Join x and y?
Error in `left_join()`:
! `by` must be supplied when `x` and `y` have no common variables.
ℹ Use `cross_join()` to perform a cross-join.Looks like we need to specify by (our 🔑)
Error in `left_join()`:
! Join columns in `y` must be present in the data.
✖ Problem with `spp_code`.Still not working!
How Do We Join x and y?
Error in `left_join()`:
! `by` must be supplied when `x` and `y` have no common variables.
ℹ Use `cross_join()` to perform a cross-join.Looks like we need to specify by (our 🔑)
Error in `left_join()`:
! Join columns in `y` must be present in the data.
✖ Problem with `spp_code`.Still not working!
Applications of data joins in forestry
Today: Applying Allometric Equations Using Joins
Allometric equations are regression equations that relate measurements like species, DBH, and perhaps height to more difficult and expensive to measure quantities such as stem volume or biomass.
Today: Applying Allometric Equations Using Joins
Allometric equations are regression equations that relate measurements like species, DBH, and perhaps height to more difficult and expensive to measure quantities such as stem volume or biomass.
- Allometric equations take the inexpensive measurements as input (e.g., species and DBH) and return estimates of the expensive quantity (e.g., height, volume, biomass)
Today: Applying Allometric Equations Using Joins
Allometric equations are regression equations that relate measurements like species, DBH, and perhaps height to more difficult and expensive to measure quantities such as stem volume or biomass.
Allometric equations take the inexpensive measurements as input (e.g., species and DBH) and return estimates of the expensive quantity (e.g., height, volume, biomass)
Today, we’ll focus on calculating tree height based on DBH and species.
The Model
The model we’ll use to calculate height is as follows \[ \text{height} = 4.5 + \exp\left( \beta_1 + \frac{\beta_2}{\text{DBH} + 1.0} \right) \] where \(\beta_1\), \(\beta_2\) are stored in “datasets/FVS_NE_coefficients.csv” and are different for each forest vegetation code.
We’ll access data from a few tibbles in order to create our final height calculation.
The Data: stands
# A tibble: 6 × 4
stand_id plot_id scientific_name DBH_in
<dbl> <dbl> <chr> <dbl>
1 1 1 Abies balsamea 11.3
2 1 1 Pinus strobus 9.8
3 1 1 Pinus strobus 10.7
4 1 3 Betula papyrifera 15.4
5 1 3 Pinus strobus 13.1
6 2 1 Larix laricina 7.1[1] 15 4The Data: stands
# A tibble: 6 × 4
stand_id plot_id scientific_name DBH_in
<dbl> <dbl> <chr> <dbl>
1 1 1 Abies balsamea 11.3
2 1 1 Pinus strobus 9.8
3 1 1 Pinus strobus 10.7
4 1 3 Betula papyrifera 15.4
5 1 3 Pinus strobus 13.1
6 2 1 Larix laricina 7.1[1] 15 4Big Picture: We’d like to calculate tree height for each row of this tibble. To do so, we’ll need to load another tibble.
The Data: FVS Coefficients (our \(\beta\)’s)
# A tibble: 108 × 3
USFS_FVS_code beta_1 beta_2
<chr> <dbl> <dbl>
1 BF 4.51 -6.01
2 TA 4.51 -6.01
3 WS 4.51 -6.01
4 RS 4.51 -6.01
5 NS 4.51 -6.01
6 BS 4.51 -6.01
7 PI 4.51 -6.01
8 RN 4.51 -6.01
9 WP 4.61 -6.19
10 LP 4.69 -6.88
# ℹ 98 more rowsQ: How can we join these tibbles?
# A tibble: 6 × 4
stand_id plot_id scientific_name DBH_in
<dbl> <dbl> <chr> <dbl>
1 1 1 Abies balsamea 11.3
2 1 1 Pinus strobus 9.8
3 1 1 Pinus strobus 10.7
4 1 3 Betula papyrifera 15.4
5 1 3 Pinus strobus 13.1
6 2 1 Larix laricina 7.1# A tibble: 6 × 3
USFS_FVS_code beta_1 beta_2
<chr> <dbl> <dbl>
1 BF 4.51 -6.01
2 TA 4.51 -6.01
3 WS 4.51 -6.01
4 RS 4.51 -6.01
5 NS 4.51 -6.01
6 BS 4.51 -6.01A: We need another tibble!
A: We need another tibble!
The “spp_codes” tibble contains the columns needed to be able to link “stands” and “ht_coeffs”.
# A tibble: 6 × 5
common_name scientific_name USFS_FVS_code USFS_FIA_code PLANTS_code
<chr> <chr> <chr> <dbl> <chr>
1 balsam fir Abies balsamea BF 12 ABBA
2 tamarack Larix laricina TA 71 LALA
3 white spruce Picea glauca WS 94 PIGL
4 red spruce Picea rubens RS 97 PIRU
5 Norway spruce Picea abies NS 91 PIAB
6 black spruce Picea mariana BS 95 PIMA Now We Can Join Our Data
# A tibble: 3 × 4
stand_id plot_id scientific_name DBH_in
<dbl> <dbl> <chr> <dbl>
1 1 1 Abies balsamea 11.3
2 1 1 Pinus strobus 9.8
3 1 1 Pinus strobus 10.7stands <- stands %>%
left_join(spp_codes, by = "scientific_name") %>%
select(-common_name, -USFS_FIA_code, -PLANTS_code) # remove unecessary columns
head(stands, n = 3)# A tibble: 3 × 5
stand_id plot_id scientific_name DBH_in USFS_FVS_code
<dbl> <dbl> <chr> <dbl> <chr>
1 1 1 Abies balsamea 11.3 BF
2 1 1 Pinus strobus 9.8 WP
3 1 1 Pinus strobus 10.7 WP - Now, “stands” contains the USFS_FVS_code column
Q: Why did we use a left_join() in the last slide?
Q: Why did we use a left_join() in the last slide?
A: We want to retain all the rows of “stands”, but only the rows in “spp_codes” that match in “stands”.
Q: How can we check if all rows had a scientific_name match?
Q: How can we check if all rows had a scientific_name match?
A: Use anti_join()!
Q: How can we check if all rows had a scientific_name match?
A: Use anti_join()!
Join \(\beta\)’s to “stands”
Join \(\beta\)’s to “stands”
Q: What type of join should we use?
Join \(\beta\)’s to “stands”
Q: What type of join should we use?
A: A left_join() (or a right_join(), if you’d like)
stands <- left_join(stands, ht_coeffs, by = "USFS_FVS_code")
# or alternatively
# stands <- right_join(ht_coeffs, stands, by = "USFS_FVS_code")
head(stands)# A tibble: 6 × 7
stand_id plot_id scientific_name DBH_in USFS_FVS_code beta_1 beta_2
<dbl> <dbl> <chr> <dbl> <chr> <dbl> <dbl>
1 1 1 Abies balsamea 11.3 BF 4.51 -6.01
2 1 1 Pinus strobus 9.8 WP 4.61 -6.19
3 1 1 Pinus strobus 10.7 WP 4.61 -6.19
4 1 3 Betula papyrifera 15.4 PB 4.44 -4.09
5 1 3 Pinus strobus 13.1 WP 4.61 -6.19
6 2 1 Larix laricina 7.1 TA 4.51 -6.01Now We Can Make Our Calculation!
Recall the equation for height: \[ \text{height} = 4.5 + \exp\left( \beta_1 + \frac{\beta_2}{\text{DBH} + 1.0} \right) \]
Now We Can Make Our Calculation!
Recall the equation for height: \[ \text{height} = 4.5 + \exp\left( \beta_1 + \frac{\beta_2}{\text{DBH} + 1.0} \right) \]
stands <- stands %>%
mutate(height_ft = 4.5 + exp( beta_1 + beta_2 / (DBH_in + 1) ) ) %>%
select(-beta_1, -beta_2) # Don't need these anymore
head(stands)# A tibble: 6 × 6
stand_id plot_id scientific_name DBH_in USFS_FVS_code height_ft
<dbl> <dbl> <chr> <dbl> <chr> <dbl>
1 1 1 Abies balsamea 11.3 BF 60.2
2 1 1 Pinus strobus 9.8 WP 61.1
3 1 1 Pinus strobus 10.7 WP 63.6
4 1 3 Betula papyrifera 15.4 PB 70.5
5 1 3 Pinus strobus 13.1 WP 69.2
6 2 1 Larix laricina 7.1 TA 47.7Next time
- Reshaping data with
tidyr